Resources
Everything you might need. In one page.
Management of drug–drug interactions of targeted therapies for haematological malignancies and triazole antifungal drugs
Roger J Brüggemann, Rebecca Verheggen, Emmy Boerrigter, Marta Stanzani, Paul E Verweij, Nicole M A Blijlevens, Russell E Lewis
Over the past 10 years, the number of targeted therapies for haematological malignancies has substantially increased, and many new drugs have entered the market. Many of these therapies have shown improved disease-free survival and reduced toxicity compared with existing treatments, especially in older patients. However, most of these new drugs undergo extensive hepatic metabolism and exhibit moderate to severe drug–drug interactions with triazole antifungal agents, which are essential for the prophylaxis and long-term treatment of invasive fungal infections. In this Review, we give a comprehensive overview of all known drug–drug interactions between new targeted drugs for haematological malignancies and antifungal drugs, in particular the triazoles. We begin with a general background on drug–drug interactions. Next, we provide a management strategy for the use of each targeted haematological drug, and discuss the possible role of therapeutic drug monitoring for both the triazole and the haematological drugs. This Review aims to provide practical guidance to clinical haematologists on managing the complex interplay between targeted therapies for haematological malignancies and triazole antifungal drugs, to pursue better outcomes for their patients.
Introduction
Over the past two decades, targeted therapies have begun to replace traditional cytotoxic chemotherapy for many malignancies. These therapies have brought substantial advances in terms of overall survival and tolerability, especially in older patients or those who are frail. However, some of these drugs might be associated with unique risks of developing invasive fungal infections.
Fungal infections are a severe threat to overall survival in patients with haematological malignancies, and affect the dose intensity or the timing of life-saving chemotherapy or haematopoietic stem-cell transplant- ation. Aspergillus-related infection is most frequently observed, followed by Candida infections;1 all such infections need immediate treatment. According to the latest guidelines,2,3 triazole antifungals are the drugs of choice for the treatment and prophylaxis of both Aspergillus and Candida infections.
The inherent problem with targeted therapies is that most of the drugs used are metabolised by cytochrome P450 (CYP) enzymes in the liver. This process predisposes the drugs to potentially severe pharma- cokinetic drug–drug interactions, especially when used with triazoles (voriconazole, posaconazole, itraconazole, fluconazole, and isavuconazole), which are moderate-to- strong inhibitors4,5 of CYP3A4. Unfortunately, clear clinical guidance on how to manage these interactions is scarce for many new targeted therapies, leaving clinicians to choose between the potential risks of reducing doses of the effective targeted therapy, avoiding the use of effective (oral) prophylaxis or the treatment of infections with triazoles, or accepting an increased risk of potentially severe toxic effects if no treatments are adapted. Because these drug interactions represent a clear danger to the successful treatment of both fungal infections and the underlying malignancy, this Review aims to provide the latest overview of the presence or absence of drug–drug interactions between targeted drugs for haematological malignancies and antifungal drugs. We have attempted to provide clinical recommendations on how to manage drug–drug interactions by adapting therapy and monitoring treatment.
Mechanism of drug interactions
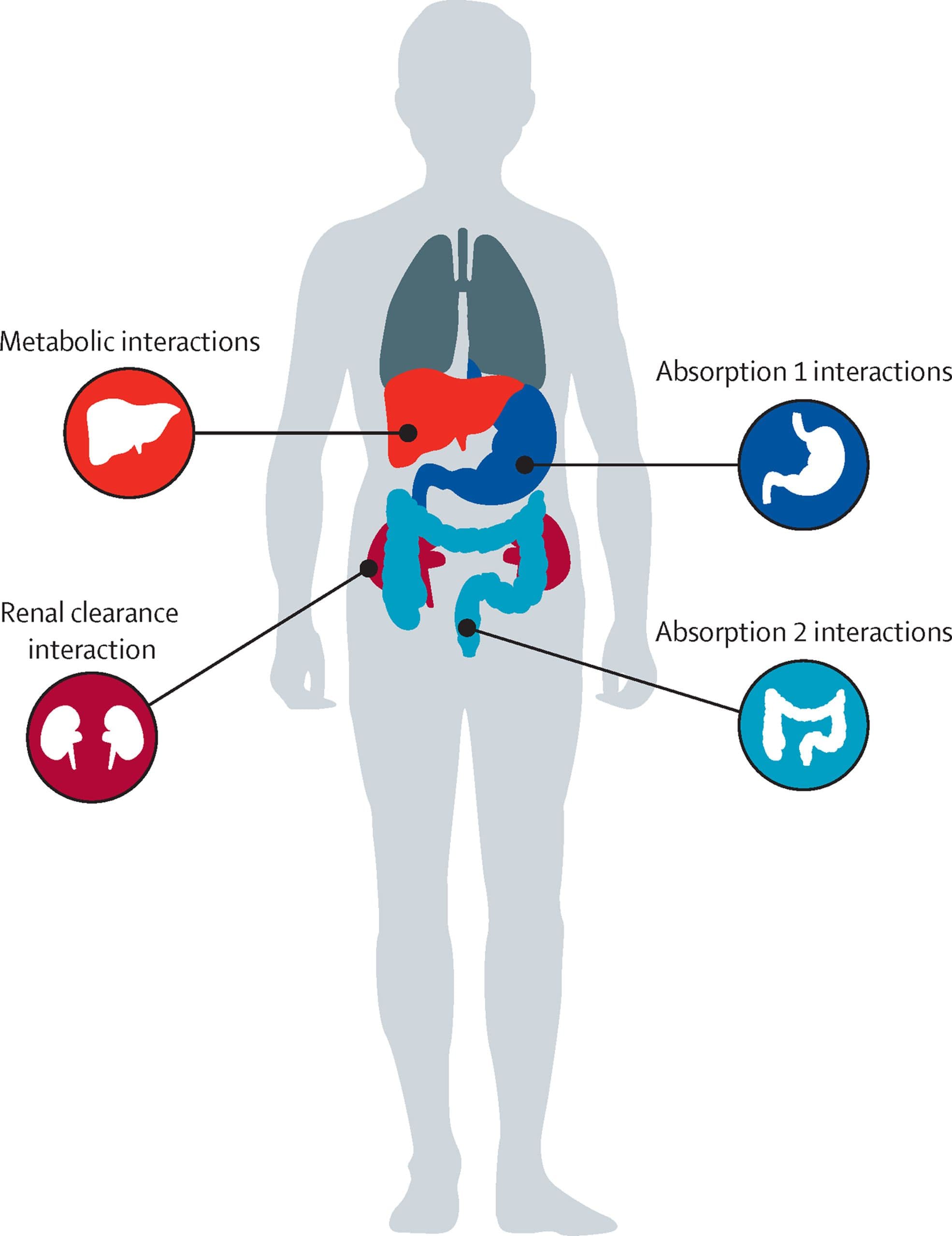
In general, drug interactions can be classified as either pharmacokinetic or pharmacodynamic. Pharma- cokinetic interactions occur at the level of drug absorption, distribution, metabolism, and elimination (figure). Pharmacodynamic interactions are defined as one drug influencing the pharmacological effect of the other. In this setting two drugs, for instance, compete for the same target, thereby causing either a synergistic effect or an antagonistic effect, resulting in excessive toxicity, enhanced therapeutic response, or a diminished clinical effect. With respect to the interaction between targeted drugs for haematological malignancies and antifungal drugs, our focus will be on pharmacokinetic interactions.
Drug interaction terminology
In terms of drug–drug interactions, a drug can be either a perpetrator or a victim. The perpetrator causes the drug interactions that affect the pharmacokinetic behaviour of the victim. In the case of a bidirectional drug–drug interaction, a single drug is both the perpetrator and the victim. Perpetrator drugs can be either inhibitors or inducers. Inhibition involves the direct (competitive) or indirect (non-competitive) blocking of a selected metabolising enzyme, and the effect can be either reversible or irreversible. In the direct setting, a drug mimics the natural substrate, thereby competing for binding to the active site of the enzyme. In a non-competitive interaction, the perpetrator drug binds to the enzyme at a site other than the active site. As a consequence, the shape of the enzyme is altered, and the normal substrate can no longer bind to the active site. After clearance of the perpetrator drug, typically 4–5 times its half-life, the inhibition is no longer present. The effect of inhibition on the suppression of gene function or on antimetabolites is beyond the scope of this Review and will not be discussed here. Victim drugs are classified as being sensitive or moderately sensitive. Sensitive index substrates are drugs that show an increase in area under the concentration–time curve (AUC) of five times or more with strong index inhibitors of a given metabolic pathway, whereas moderately sensitive substrates show an increase in AUC between two or more and less than five times. Inhibitor drugs are classified according to the magnitude by which they increase the AUC of sensitive index substrates of a specific metabolic pathway: five times or more (strong), between two or more and less than five times (moderate), and between 1·25 or more and less than 2 times (weak [US Food and Drug Administration (FDA)] or mild [European Medicines Agency (EMA)]).
Whereas the effect of inhibition is instantaneous, induction takes time to reach its maximum effect. Induction results in increased drug metabolism due to the upregulation of enzyme activity, which itself results from increased protein biosynthesis. After drug clearance, the inhibitive effect no longer occurs, whereas the inductive effect takes longer to stop. The half-life of the enzyme—after clearance of the perpetrator drug— establishes how long the inductive effect persists, and in some cases might take several weeks before the effect is lost.
As a result of pharmacokinetic drug interactions, both the on-target and off-target effects of a victim drug can be altered. An on-target effect is an exaggerated therapeutic effect at the target, whereas off-target effects are caused by binding of the drug to an unintended target, and most frequently result in unwanted side-effects. The off-target effect can be biologically related to the target of interest, but is often unrelated.
Changes in on-target and off-target effects can be driven not only by the parent compound, but also by its formed metabolites. The pharmacokinetics of both could be altered due to a pharmacokinetic drug interaction, with changes in both on-target and off-target effects; however, the off-target effects of metabolites are rarely studied in the clinical setting.
Pharmacokinetic drug interactions
Drug interactions at the level of absorption
Although most previous anticancer drugs were admin- istered intravenously, many newer drugs, including most of those in this Review, can be administered orally. As such, changes in absorption of these drugs might have a major role in their activity. Before reaching the systemic circulation, drugs must be absorbed and pass through the mucosal lining of the digestive tract. The absorption of a drug, and therefore its pharmacokinetic profile, can be influenced by the presence or absence of food and by the gastric pH. The effect of food and gastric pH is dependent on the pharmaceutical formulation of the drug (eg, instant release, delayed release, and coated), its physicochemical properties, and the amount and content of food (eg, low fat or high fat). The motility of the gastrointestinal tract might also have a role. Some of the targeted haematological drugs that we discuss here can be affected by the presence of food; however, a detailed discussion of such interactions is beyond the scope of this Review. Similar to the targeted haematological drugs, some triazoles are also affected by food. The reduced absorption of antifungal drugs in the presence of food will lead to lower exposure to the perpetrator drug, resulting in a less pronounced effect, typically inhibition, on the victim drug.
Several other processes in the intestinal mucosal system have important effects on the oral bioavailability of drugs. Drugs can be either passively transported or require active transport via a multitude of drug transporters, including both influx (organic cation or anion) or efflux (eg, P-glycoprotein or breast cancer resistance protein) transporters. The intestinal epithelial cells also contain CYP enzymes—predominantly CYP3A4—which results in presystemic clearance of the selected drug if it is a substrate of these enzymes. The introduction of a perpetrator drug that blocks CYP3A4 activity will result in reduced clearance of, and therefore higher systemic exposure to, the victim drug. Similarly, the inhibition of P-glycoprotein, which can be achieved by the co— administration of the triazole drugs itraconazole and posaconazole, and to a lesser extent isavuconazole,9 blocks the efflux of a victim drug to the luminal site, therefore retaining more of the drug in the systemic circulation, resulting in higher exposures. The effect on presystemic clearance is characteristically important when switching from intravenous to oral therapy and vice versa; however, this issue is of little relevance as most targeted haematological drugs are only available as oral drugs.
Metabolic drug interactions
The dominant site of drug metabolism is the liver, where two major types of metabolic reaction occur: phase I reactions, which involve oxidation, reduction, and hydrolysis; and phase II reactions, which involve the conjugation of drugs. These reactions are mediated by the CYP enzyme system. Of the phase I enzymes, CYP3A4 is the most abundant, followed by CYP2C9, whereas CYP2A6, CYP2D6, CYP2B6, and CYP2C19 are less abundant.10 The effect of these metabolic reactions on the concentration of the drugs involved will establish the magnitude of the metabolic drug–drug interactions. A given drug can be a substrate of several CYP enzymes. Anticipating the pharmacokinetics of a drug becomes more complex if the drug can be involved in multiple metabolic pathways, or if it has an active metabolite.
The affinity for phase I or II enzyme systems is dependent on the drug. Major and minor routes of metabolism for haematological drugs, and the substrate affinities for various enzymes, are shown in table 1.
Several factors influence the expression and function of CYP enzymes, including genetic polymorphisms. These polymorphisms divide the population of enzymes into poor, intermediate, extensive, and ultrarapid metabolisers, all of which can be either homozygous or heterozygous. Both homozygous and heterozygous poor metabolisers have absent or reduced metabolic capacity, which results in higher systemic exposure to the substrate drug due to low metabolic clearance. Unlike poor and intermediate metabolisers, ultrarapid metabolisers show a gain of function in terms of metabolising activity, resulting in a more rapid metabolic clearance of the substrate drug.10 Not all CYP enzymes are affected to the same extent by genetic polymorphisms, with the order of clinical importance as follows:10 CYP2D6 then CYP2C19 and CYP2A6, then CYP2B6, then CYP2C9, and finally CYP3A4 and CYP3A5.
Genetic polymorphisms of the CYP enzymes can affect both the perpetrator and the victim drug if both are substrates for the same CYP enzyme. The combination of genetic variation of CYP enzymes and drug interactions can be very challenging. For instance, voriconazole is predominantly metabolised by CYP2C19, followed by CYP3A4 and CYP2C9. If there is loss of function of CYP2C19 as a result of genetic mutations, metabolism by CYP3A4 becomes the dominant pathway (appendix p 3). As such, the effect of a CYP3A inhibitor is many times greater in patients in which the CYP2C19 pathway is blocked than in patients with normal CYP2C19 function.
In addition to genetic polymorphisms, there are several other factors that affect the function of the CYP enzyme system in drug metabolism, such as sex, age, disease state, and inflammation status.10 In addition, CYP-mediated drug– drug pharmacokinetic interactions remain one of the most important factors in the variability of drug exposure.
In recent years, knowledge of the effects of drug transporters has grown. These transporters can also play an important part in drug–drug interactions because they regulate the access of the substrate to the metabolising enzymes. Furthermore, these transporters control the influx and efflux of drugs into enterocytes and hepatocytes. Although P-glycoprotein and the hepatic uptake transporters known as organic anion- transporting polypeptides are probably the most widely investigated, there are many other relevant influx and efflux transporters that might have a role in drug–drug interactions. The guidelines on the investigation of drug interactions available from the EMA6 recommend that information on transporters involved in major elimination processes should be gained as early as possible during drug development. The need for information on the effect of drug transporters is driven by the predicted magnitude of increases in exposure to the drug after inhibition of the transporter, the clinical consequences of which are obviously important. Relevant in-vitro experiments have been done for all targeted haematological drugs that we review here, and data from these studies are summarised in table 1. In-vitro data are only provided if they have possible clinical relevance or if their clinical relevance is currently unknown.
Pharmacodynamic drug interactions
Pharmacodynamic interactions between antifungal drugs and targeted haematological drugs can be additive (the total effect is equal to the sum of the effect of each individual drug), synergistic (the total effect of the combination is greater than the sum of the effects of the individual drugs), or antagonistic (the total effect of the combination is less than then the sum of the effects of the individual drugs). Because the mechanisms of action of antifungal drugs and targeted haematological drugs do not overlap, any pharmacodynamic interactions between these drugs will result from an overlap in toxicity profiles. Triazole drugs are known for their hepatotoxicity and their propensity to induce QT prolongation, both of which could be exacerbated by the combination of triazoles with haematological drugs.
In the appendix (p 1), we state whether there is a known risk of the targeted haematological drugs evoking QT prolongation. Combining the haematological drugs that are associated with QT prolongation with voriconazole, posaconazole, and itraconazole will result in an additive effect. The only exception within the class of triazoles is isavuconazole, which shows QT shortening rather than QT prolongation.12 However, whether the combination of a QT-shortening drug and a QT-prolonging drug reduces the QT-prolongation effect is unclear; if this is the case, isavuconazole might be a preferred choice in combination with haematological drugs that prolong QT intervals.
When do drug interactions become clinically relevant?
Although many targeted haematological drugs are prone to drug–drug interactions with triazoles, the clinical relevance of the interaction depends on numerous factors. Perpetrator drugs can have different potencies, which are well described for the triazole class. Of the triazoles used for the prophylaxis or treatment of mould disease, itraconazole is the most potent CYP3A4 inhibitor, followed by voriconazole and posaconazole. Isavuconazole and fluconazole are the weakest inhibitors (appendix p 3). The FDA7 provides a comprehensive overview of substrates, inhibitors, and inducers, including clinical index drugs for use in drug–drug interaction studies. Knowledge of the effect of the perpetrator drug on the victim drug is therefore crucial to assess whether dose adjustments of either drug are needed, or whether their concomitant administration should be avoided altogether.
In table 2 we provide an overview of all available published clinical evidence to date, to the best of our knowledge, on drug–drug interaction studies between triazole antifungal drugs and targeted haematological drugs. The table also includes the direction and extent of the effects of individual triazole drugs on the haematological drugs. In cases for which clinical drug– drug interaction studies are absent, we use information from physiological-based pharmacokinetic modelling to mathematically predict the extent of the expected interaction. Previous knowledge of the inhibitory potency and physiochemical properties of the relevant drugs is used to make these predictions. In the appendix (p 2), we present an overview of the effect sizes of drug–drug interactions based on in-silico predictions.
If the victim drug has a narrow therapeutic range, its drug–drug interactions tend to be more clinically relevant, because increased therapeutic drug exposure means that the toxicity threshold of the drug is rapidly reached. The therapeutic range of a drug is defined as the concentration range in which an optimal clinical effect is achieved. If the concentration of the drug increases above the therapeutic range, patients might encounter adverse effects. Examples include the haematological drugs glasdegib, bosutinib, and venetoclax (table 2). When each of these drugs is combined with a triazole, the concentration of the haematological drug increases beyond the therapeutic range, which can lead to persistent neutropenia and cytopenia. Conversely, although drugs with a wide therapeutic range, such as imatinib and midostaurin, might also be prone to considerable increases in parent drug exposure as a result of drug–drug interactions, these increases are less clinically relevant as the augmented exposure does not result in clinically important toxic effects (table 2).
Many targeted haematological drugs are metabolised through multiple pathways (table 1). If one of these pathways is blocked owing to the presence of a perpetrator drug, alternative metabolic routes might become more important. However, if a perpetrator drug blocks multiple pathways, there is no escape pathway available. For example, if ruxolitinib is combined with posaconazole (an inhibitor of CYP3A4), the CYP3A4 route will be blocked but the CYP2C9 route will remain available. However, if ruxolitinib is combined with fluconazole (a mild inhibitor of CYP3A4 and a strong inhibitor of CYP2C9) both the CYP3A4 and CYP2C9 routes will be blocked. The exposure to ruxolitinib is greater when it is combined with fluconazole than when it is combined with posaconazole.32 In addition, blocking a primary route or inducing a minor or secondary route of metabolism by a perpetrator could result in diversion of the victim drug into an alternative pathway. This blockage of the primary route or the induction of secondary routes could then result in the generation of other (toxic) metabolites, or change the overall activity of the drug if active metabolites have an important role in antitumour efficacy or have substantial off-target effects.
The management of drug–drug interactions in daily clinical practice is often not suited to a generalised approach, and personalised interventions are usually required. An awareness of the mechanism of interaction, and an understanding of the extent of the interactions and the effect on metabolites, is essential for the provision of optimal and personalised treatment when targeted haematological drugs are given concomitantly with triazoles. Table 3 provides the haematologist with suggested clinical guidance, for which we translate the knowledge we have discussed into a clinical management strategy that can be used in everyday practice. The advice provided might differ from the summary of product characteristics of the targeted drug. Because not all available information finds its way to the label, the pharmaceutical industry generally does not provide recommendations outside the legally documented label information. In table 3, we translate all current evidence and clinical knowledge of safety into suggested practical recommendations.
Following the review of drug–drug interactions, we next discuss how therapeutic drug monitoring can be used to guide and safeguard optimal therapy.
Therapeutic drug monitoring of targeted therapies
There are many reasons why routine therapeutic drug monitoring can improve the safety and efficacy of targeted haematological drugs, such as oral smallmolecule kinase inhibitors (SMKIs), and the management of drug–drug interactions.33 SMKIs are known to show high interindividual pharmacokinetic variability.34 For some of these agents, lower-than-average serum or plasma drug exposures are associated with poorer cytogenetic tumour response and higher rates of malignancy progression.35 Conversely, higher-thanaverage drug exposures, especially in the case of drug– drug interactions with CYP inhibitors, such as triazole antifungals, are associated with a greater risk of treatment-limiting drug toxicities. By detecting changes in the exposure of the patient to the targeted chemotherapy, and possibly to metabolites with or without on-target and off-target effects, if present, therapeutic drug monitoring could enable more precise and individualised dosing adjustments with concomitant triazole antifungal therapy, therefore enabling a greater proportion of patients to continue both life-saving therapies in combination.
However, evidence of the clinical utility of routine therapeutic drug monitoring in patients receiving SMKIs for haematological malignancies is still preliminary, as it is more common in the treatment of solid tumours.34,35 Most studies that have evaluated therapeutic drug monitoring for SMKIs have focused on confirming or improving exposures of SMKIs rather than the management of drug interactions.35 We anticipate an increase in interest in the use of therapeutic drug monitoring in haematology. It can be used to guide drug dosing with more precision, and aligns with the goal of targeted and individualised therapy. For patients who have already had considerable toxic effects from SMKI therapy, therapeutic drug monitoring could help to differentiate between adverse effects that occur during potentially high drug exposures (which could potentially be managed by reducing treatment doses or stopping the administration of the interacting drug) and toxic effects occurring during average or low SMKI exposures, in which case a reduction in dose could place the patient at an unacceptable risk of potentially fatal disease progression.35 It can also be used to show non-adherence to a treatment plan.
The relationship between plasma drug exposures and surrogate pharmacodynamic biomarkers has been shown for only a few SMKIs (table 4). The measurement of trough concentrations of these SMKIs, when the steady-state concentration has been reached, can be used to confirm adequate drug exposures for reaching and maintaining a cytogenetic or molecular response. The clinical implementation of routine therapeutic drug monitoring for targeted therapy can also present several logistical challenges. Onsite drug assays for rapid therapeutic drug monitoring might be available in many treatment centres; however, resources for assay validation, rapid test reporting, and expert clinical interpretation are still required to enable the translation of results into an optimal clinical decision.33,64 One randomised study found that the time to development of severe adverse events, treatment discontinuation, admission to hospital, or death in patients taking oral SMKIs was reduced (hazard ratio 0·48; 95% CI 0·32–0·71; p<0·001) in patients who had structured consulting visits and continued follow-up with clinical pharmacists.65 Knowledge of patient adherence, the optimal timing of oral SMKI therapy with respect to consuming meals, and important drug–drug interactions with non-triazole therapies (including nonprescription medications and herbal remedies available over the counter) is necessary to correctly interpret the therapeutic drug monitoring results.65 Therapeutic drug monitoring is therefore only one component of a multistep, integrated process for managing chronic SMKI therapy in patients who require antifungal therapy.
The simplest approach for incorporating therapeutic drug monitoring into the monitoring of targeted therapy is to measure a trough concentration of the targeted agents at steady state to estimate drug exposure and clearance, then use an established algorithm or nomogram to adjust low or excessive exposures by varying the fixed dose. However, because trough concentrations are required to generate a nomogram, measurements must be taken at fixed times, which can be an issue in terms of outpatient monitoring. In addition, dosing adjustments might be not be precise enough if the patient is not well represented by the population that was used to derive the nomogram.66 Bayesian approaches that incorporate both information on relevant population pharmacokinetic (non-linear effects) models and information on exposure and efficacy and toxicity responses from previous clinical trials could improve the precision of personalised dose adjustments of SMKIs. These models are used in two ways. First, the concentration–time profiles of SMKI drugs can be predicted a priori on the basis of the patient’s clinical characteristics (eg, sex, age, and weight) with a relevant pharmacokinetic model. Second, the predicted drug exposures can then be confirmed a posteriori by direct measurement of plasma drug concentrations, enabling model parameters to be adjusted to fit the patient-specific pharmacokinetic profile and allowing for more precise dose adjustments when treatment with interacting triazole drugs is started or stopped.66 The use of Bayesian models to inform dosing also enables greater flexibility in the timing of blood samples for calculating patient-specific pharmacokinetic parameters, provided that actual sampling times are documented. Unfortunately, only a few user-friendly, web-based Bayesian dosing programmes for SMKIs are available, such as InsightRX or have undergone European certification or regulatory approval. Therefore, their use in dosage adjustment and the management of antifungal drug interactions remains at an investigational stage.
Alternative prophylaxis strategies
Because nearly all targeted therapies for haematological malignancies are metabolised by CYP3A4, antifungals that do not inhibit CYP enzymes, such as echinocandins or liposomal amphotericin B, could be considered as alternatives to triazoles in patients who require antifungal prophylaxis or treatment. These intravenous-only antifungals can be a useful temporary solution for antifungal treatment, but they have many limitations for chronic outpatient therapy in terms of convenience and patient safety. Intermittent dosing regimens for both echinocandins and liposomal amphotericin B have been explored for prophylaxis in adult and paediatric patients;67–69 however, the relative effectiveness of these infrequent dosing regimens compared with established oral triazole prophylaxis regimens is still unproven.
The use of non-triazole drugs in antifungal prophylactic therapy might also be associated with a higher risk of breakthrough fungal infections. According to the European Society of Clinical Microbiology and Infectious Diseases guidelines2 for the treatment of invasive aspergillosis, echinocandin monotherapy is not recommended, and some observational studies have reported higher rates of breakthrough infection with echinocandin treatment than with mould-active triazole prophylaxis following induction chemotherapy for acute myeloid leukaemia or myelodysplastic syndrome.70,71 Fulminant breakthrough infections with rare (non-Candida and non-Cryptococcus) yeast have also been reported, and are associated with high rates of morbidity and mortality.72,73 We emphasise that the use of a triazole alongside an informed dose modification of the targeted haematological drug might be the preferred strategy to avoid life-threatening fungal infections. An example of such a strategy could be the fixed combination of posaconazole and venetoclax (dose reduced by at least 75% to a maximum of 25% of the regular dose).
If a mould-active triazole antifungal is the only option for treatment or prophylaxis, isavuconazole has several advantages over voriconazole or posaconazole. Unlike other triazoles, isavuconazole is reported to not prolong the corrected QT (QTc) interval.12 Compared with voriconazole or posaconazole, isavuconazole is also a less potent inhibitor of human CYP3A4, 7 and it has fewer cutaneous, hepatic, and visual toxic effects than voriconazole.74 However, several observational studies have reported higher rates of breakthrough invasive fungal infection in patients receiving isavuconazole, and such findings require confirmation in larger trials.75,76 The half-life of isavuconazole is longer than that of other triazoles (130 h), which will be of concern if a patient has severe toxic effects. Furthermore, observational studies in patients who received mould-active triazoles with the BCL-2 inhibitor venetoclax reported that patients who received isavuconazole showed a longer time to neutrophil and platelet recovery than patients who received voriconazole or posaconazole.77,78 Finally, the effectiveness of isavuconazole as a prophylactic agent needs to be confirmed. Therefore, whether the risk of serious adverse events is reduced in patients that receive isavuconazole rather than the more potent CYP3A4 inhibitors voriconazole or posaconazole remains unproven.
Several new oral antifungals currently in phase 2 and phase 3 trials could have a future role in prophylaxis for patients that are receiving targeted haematological therapies.79 Rezafungin (Cidara; San Diego, CA, USA), an analogue of anidulafungin that has a longer half-life, and is currently in phase 3 trials (NCT04368559), would allow for once-weekly dosing, possibly through the subcutaneous route. Ibrexafungerp (Scynexis; Jersey City, NJ, USA) is an echinocandin-like antifungal that is available in both oral and intravenous formulations. Fosmanogepix (Pfizer; Gladstone, NJ, USA) is a broadspectrum antifungal compound that interferes with glycosylphosphatidylinositol-anchored mannoproteins in the fungal cell wall, and is being developed in intravenous and oral formulations. Both ibrexafungerp and fosmanogepix are reported to have good oral bioavailability with no clinically significant CYP3A4 inhibitory potency.80 Olorofim (F2G; Manchester, UK) is a novel inhibitor of fungal dihydroorotate dehydrogenase, an important enzyme in pyrimidine biosynthesis, which is essential for deoxyribonucleic acid synthesis. The compound is currently being studied as an oral formulation for the treatment of invasive aspergillosis and other rare mould infections,81 and is reported to be a weak inhibitor of CYP3A4. Finally, several of the nextgeneration tetrazoles (VT-1129, VT-1161 [also known as oteseconazole], and VT-1598) are in earlier stages of clinical development. By replacing the metal-binding triazole with a tetrazole, greater selectivity for the fungal lanosterol 14α demethylase (CYP51) versus human CYP3A4 can be achieved,82,83 which should theoretically improve antifungal potency and reduce the risk of serious drug–drug interactions with targeted haematological therapies.
Future considerations
Many of the targeted drugs for haematological malignancies show clinically significant drug–drug interactions. In this Review, we have focused on the interactions of these targeted drugs with antifungal agents, and the clinical consequences of these interactions and their subsequent management. There are also many other interactions of targeted haematological drugs that are of clinical relevance, such as the interactions with letermovir.
Drug–drug interactions are a threat to the management of patients undergoing haematological treatments, with the risk that the therapy could be unsuccessful or could result in excessive toxic effects. To ensure the safe and adequate use of targeted drugs for haematological malignancies, knowledge of the pharmacological background of drug interactions and the risk of either subtherapeutic or supratherapeutic exposure is crucial. The management of drug–drug interactions requires a multidisciplinary approach. Increased awareness of potential drug–drug interactions is key to minimising the risk. Haematologists could liaise with pharmacologists to address this challenge, as many drug–drug interactions remain unresolved and require a hypothetical translation from other drugs.
In the very near future, we might even take advantage of drug–drug interactions between targeted drugs for haematological malignancies and triazoles. Fixed combinations of these two drug classes could prevent fungal infections from occurring while simultaneously maintaining maximal efficacy of the haematological treatment. Such an approach would even reduce the cost of the targeted haematological treatment, as a smaller amount of the targeted drug would be required because of the boosting effect of the triazole. Prospective studies should show that such fixed-combination, triazoleboosted targeted therapies are as effective against haematological malignancies as the targeted drug alone, while also providing suitable prophylaxis for mould infections.
Finally, therapeutic drug monitoring and pharmacometric analysis are already available in haematology, and can be used in the clinical setting immediately. We anticipate major advances in the field of targeted drugs for haematological malignancies, particularly because both the pharmacokinetics and pharmacodynamics can be linked to guide optimal exposure. Targeted therapy will then become truly targeted.